





TOP LEFT: Albino Squirrel. Note the pink eyes. From Brooker, R.J. "Genetics", Addison Wesley Longman, 1999, p 623.
TOP RIGHT: Albino Wildebeest. Note that this individual is very young. Because of their lack of protective coloration, albinos in most species do not survive long. From Brooker, R.J. "Genetics", Addison Wesley Longman, 1999, p 623.
MIDDLE LEFT: Albino Sea Turtle. http://ankur.netfx.net/pics.html
MIDDLE RIGHT: A Hopi girl with albinism. From Weaver R.F. and Hedrick, P.W.; "Genetics", WC Brown., 1997, p 36.
BOTTOM LEFT: Human family in which albinism is inherited. From Brooker, R.J. "Genetics", Addison Wesley Longman, 1999, p 623.
BOTTOM RIGHT: Albino Burmese
Python. http://www.amberle.com/ats/
|
|
|
|
|
|
The set of six pictures above shows the phenotype of Albinism. Albinos do not produce the pigment melanin. This is generally a dark pigment, but there are different forms with different colors. Melanin is deposited in the skin, hair and irises of the eyes. It accounts for:
As with so many other genetic diseases the
explanation lies in biochemistry. Most of the biological
chemicals upon which life depends (sugars, amino acids, DNA bases, lipids
etc) must be chemically synthesized by cells. The series of
chemical reactions used by cells to synthesize biological chemicals (called
a biosynthetic pathway)
are exactly the same reactions used by organic chemists in a laboratory.
The only difference is that cells use enzymes
to catalyze these reactions rather than heat, heavy metals or pH adjustments.
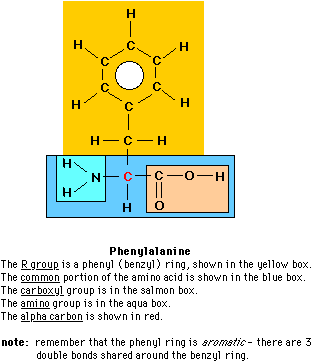
Albinism results from defects in the the biosynthetic pathway leading from the amino acid phenylalanine. Before you go farther view the chemical structure of phenylalanine and the 3-dimensional shape of phenylalanine
The phenyalanine pathway is involved in 3 important genetic diseases or conditions of humans:
| Each of these genetic diseases results from a defective protein - one of the enzymes which catalyzes a critical step in the biochemical pathway of phenylalanine. You will remember from Principles of Biology I and Cell Biology that enzymes function because a substrate fits into their active site. This means that the substrate must fit into the active site - if it cannot fit, then the enzyme cannot facilitate the reaction. In these 3 genetic diseases, the SHAPE of the enzyme is defective. Because of the defective shape, the substrate cannot fit into the active site and the reaction is not catalyzed. |
Tyrosine is then
hydroxylated a second time to produce 3,4-dihydroxyphenylalanine
(DOPA). The enzyme which catalyzes this second hydroxylation is tyrosine
hydroxylase. DOPA is the precursor
in a sequence of many chemical reactions which ultimately produce the brown
pigment melanin (the biochemical pathway has been simplified,
and appears to indicate that melanins is produced directly from DOPA;
this is not so). The first reaction in this series of reactions is
catalyzed by an enzyme called tyrosinase.
Albinism results when either of two enzymes - tyrosine hydroxylase or tyrosinase - is defective! They are defective in having the wrong SHAPE, so that their substrates cannot fit into the active site.
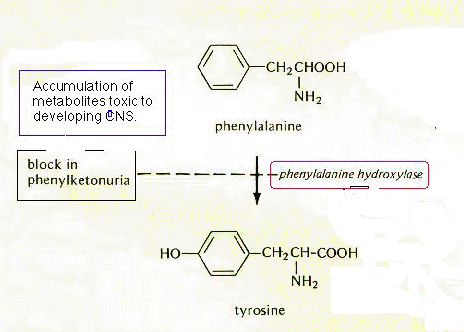
Phenylketonuria occurs when phenylalanine hydroxylase doesn't work. If the shape of phenylalanine hydroxylase is defective, then the substrate phenylalanine cannot fit in and the product tyrosine is not synthesized. Unlike tyrosine, there is no good way for cells to get rid of excess phenylalanine. Consequently, if phenylalanine is not converted to tyrosine, it does accumulate in cells and the levels in the bloodstream go up. When it reaches high levels phenylalanine is metabolized, but these products are toxic to developing cells in the brain and central nervous system. This results in extensive damage to the brain - and mental retardation!
Another characteristic of children afflicted with PKU is their very light hair, blue eyes and fair skin. The reason for this is related to the reason for Albinism. With lower than normal levels of tyrosine, the cells of these individuals produce less DOPA and ultimately, less melanin! They are not albinos because the tyrosine hydroxylase and tyrosinase in these individuals is not defective. Some tyrosine is available in the diet and this can be used to make some melanin.
Treatment for PKU is relatively straightforward. In the U.S., all babies are tested for PKU at birth. These babies are placed on special diets low in phenylalanine until they are about 5 years old. Since the human brain is fully developed by the age of 3, the toxic products of unusual phenylalanine metabolism do not have any damaging effects past this age. However a female with PKU will have to return to the low phenylalanine diet if she becomes pregnant; otherwise the toxic metabolites will damage the immature CNS of her fetus.
About 1 in 11,000 live births are diagnosed with PKU which means that approximately 1 in 100 adults is a carrier for PKU. Of the 5500 students at Clarion, the odds are that 55 of them are carriers of the defective allele for defective phenylalanine hydroxylase which produces PKU.
The figure used to illustrate PKU was modified from Strickberger,
M.W., "Genetics 2nd ed.", Macmillan, 1976, p 601.
Alkaptonuria results when homogentisic acid oxidase does not work. This results in an accumulation of homogentisic acid to high levels. HA, like most phenolic compounds, spontaneously oxidizes to various dark colored products. These are deposited in the soft tissues of the body, especially cartilage. Hence people with alkaptonuria have very dark noses and ears. Some pigment is also deposited in the cartilage of the joints turning them black and causing arthritis in later life.
Many of the oxidized products are also excreted in the urine, so the urine is black. This forms the basis for a very simple diagnosis of the disease in newborns. Babies born with alkaptonuria (about 1 in 20,000 live births) have black diapers! Other than arthritis and esthetically unpleasing noses and ears, this genetic condition is relatively benign. Many alkaptonuriacs reproduce and survive to old age.
The figure used to illustrate Alkaptonuria was modified
from Strickberger, M.W., "Genetics 2nd ed.", Macmillan, 1976, p 601.
| RETURN to "Phenotypes" Page |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}